
Ophthalmology research in 2025 was defined by significant progress in regenerative medicine, drug de...
read more
The ophthalmology industry in 2025 continued to evolve through strategic acquisitions and partnershi...
read more
The AI in ophthalmology sector is evolving rapidly, introducing Artificial Intelligence tools in oph...
read more
The year 2024 has been a remarkable period for advancements in ophthalmology, with groundbreaking in...
read more
The year 2024 has been a pivotal one for ophthalmology, with several novel FDA approvals introducing...
read more
2024 has been a landmark year for breakthroughs in ophthalmology research, showcasing transformative...
read more
In 2024, the ophthalmic industry experienced a transformative year driven by strategic company acqui...
read more
A new analysis published in the British Journal of Ophthalmology reveals that nearly one in three ch...
read more
Lamprey Eye Disease, also known as "Lamprey Disease," is a hoax that has been circulating on the int...
read more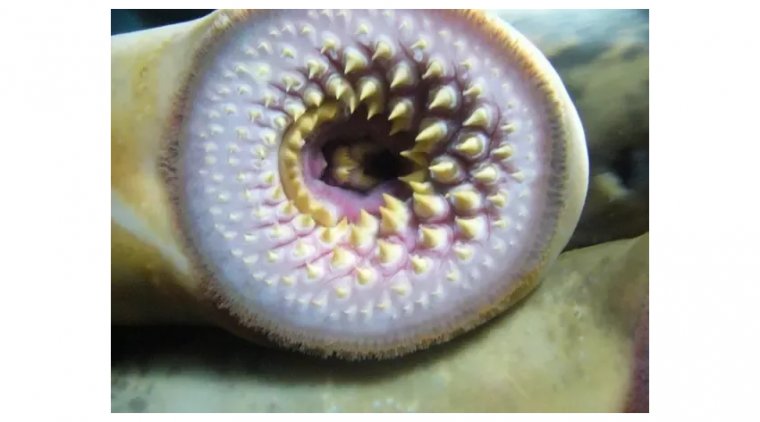
A recent study highlights the potential of ChatGPT-4.0 in accurately answering questions related to ...
read more
 More
More