(1).jpg)
How to Deal with Usher Syndrome and Retinal Degeneration
Usher syndrome is the most common condition that affects both hearing and vision; sometimes it also affects balance. The major symptoms of Usher syndrome are deafness or hearing loss and an eye disease called retinitis pigmentosa (RP).
Deafness or hearing loss in Usher syndrome is caused by abnormal development of hair cells (sound receptor cells) in the inner ear. Most children with Usher syndrome are born with moderate to profound hearing loss, depending on the type.
Less commonly, hearing loss from Usher syndrome appears during adolescence or later. Usher syndrome can also cause severe balance problems due to abnormal development of the vestibular hair cells, sensory cells that detect gravity and head movement.
Usher syndrome is a rare genetic disorder primarily characterized by deafness due to an impaired ability of the inner ear and auditory nerves to transmit sensory (sound) input to the brain (sensorineual hearing loss) accompanied by retinitis pigmentosa, a disorder that affects the retina and causes progressive loss of vision.
Researchers have identified three clinical types of Usher syndrome. The age at which the symptoms appear and the severity of symptoms that distinguishes the different types of Usher syndrome is determined by the underlying genetic cause.
- Usher syndrome is inherited as an autosomal recessive genetic trait.
- Usher syndrome is a condition characterized by partial or total hearing loss and vision loss that worsens over time.
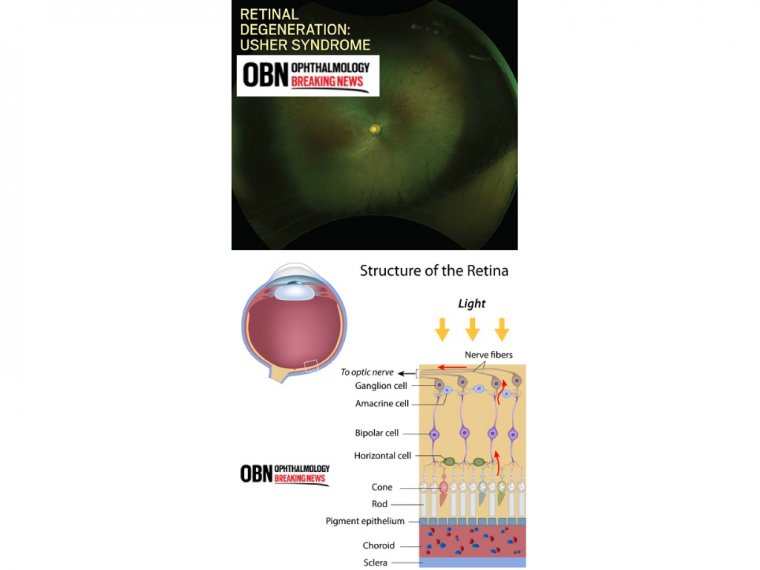
The hearing loss is classified as sensorineural, which means that it is caused by abnormalities of the inner ear. The loss of vision is caused by an eye disease called retinitis pigmentosa (RP), which affects the layer of light-sensitive tissue at the back of the eye (the retina).
Vision loss occurs as the light-sensing cells of the retina gradually break down. Loss of night vision begins first, followed by blind spots that develop in the side (peripheral) vision.
Over time, these blind spots enlarge and merge to produce tunnel vision.
Physcians who specialize in the vitreous and retina are often presented with familial conditions affecting the visual pathway.
Evaluation may involve multiple dimensions, including genealogy, genetics testing, physical examination, obtaining medical history, multimodal imaging, and electroretinography. There are numerous collaborators and colleagues who may play vital roles in their contribution.
Ultimately, it may be the duty of retina specialists to be at the center of this effort, as they are best positioned to compile all of the available details and speak in a language that patients and their family will understand.
With the advent of adenovirus viral vector (AVV) technology and its use in human embryonic kidney 293 cell lines leading to the first FDA-approved therapy for a genetic retinal degeneration, namely biallelic Leber congenital amaurosis in the form of Voretigene neparvovec-ryzl (Luxturna, Spark Therapeutics), the stakes have been raised.
While expectant management and counseling have been the norm since the first description of an inherited retinal degeneration in 1855, for the first time treatments may become available for a wide variety of conditions.
USHER SYNDROME
The condition is defined as autosomal recessive deafness (commonly congenital) with retinal findings that are clinically indistinguishable from typical retinitis pigmentosa.
Credit for discovering this condition as a unique heredity and constellation of symptoms is given to the British ophthalmologist Charles Usher.
It has been estimated that the condition’s incidence is somewhere between 1.8 and 6.2 cases per 100,000, though this figure is being refined from other reviews. About 50% of those who are born deaf and blind have Usher syndrome.
There are three clinically distinct groups whose diagnosis has been defined by the Usher Consortium. The most common is type 1, in which patients have profound sensorineural deafness and resultant pre-lingual speech impairments, vestibular symptoms, and childhood-onset retinopathy.
In type 2, there is partial and nonprogressive deafness, absent vestibular symptoms, and late retinopathy.
Type 3 results in progressive deafness starting in the second to fourth decades, adult-onset retinopathy, and hypermetropic astigmatism. It is most common for those with type 3 to be of Finnish descent.
Among the symptoms present in Usher syndrome, vestibular dysfunction is the earliest. In infancy, patients manifest a delay in motor development. Almost all children with type 1 fail to walk before the age of 18 months.
As they reach their teenage years, these same patients invariably report new symptoms or show signs of nyctalopia, whereas in types 2 and 3, these symptoms develop later in life, up to the third decade.
Visual acuity follows a similar pattern with type 1 patients developing earlier vision loss than those in type 2. Approximately 70% of those with type 1 maintained 20/200 acuity at age 40, while 95% those of a similar age group retained the same vision in type 2.
There is no increased incidence of posterior subcapsular cataract in one group more than another. Since type 3 is so rare, no such comparative studies have been conducted.
Results from electroretinography follow the above trends in visual symptoms, with type 2 being less likely to be detected as compared with type 1 in comparable age groups early in life.
As there is yet no defined or specific therapy, the role of a physician in making a diagnosis is to create an opportunity for families and patients to plan ahead by signaling the need for educational and sociopsychologic intervention to foster independence and productivity.
Additionally, cochlear implantation has been successful in allowing patients with all types of Usher syndrome to gain improved hearing. Therefore, an undiagnosed child may benefit from early detection.
Other conditions that must be considered at the time a clinician finds impaired hearing and retinitis pigmentosa-like retinal findings include: infantile Refsum disease (also called infantile phytanic acid storage disease), adult Refsum disease, Cockayne syndrome, Bardet-Biedl syndrome (BBS), Alström syndrome, Flynn-Aird syndrome, Friedreich ataxia, and Kearns-Sayre syndrome.