(1).jpg)
The Link Between Congenital Plasminogen Deficiency & Blood Ties
Two types of plasminogen deficiency are described in the medical literature. Congenital type 1 plasminogen deficiency is a quantitative disorder with parallel reductions in both the level of immunoreactive and functional plasminogen; type1 plasminogen deficiency is also called hypoplasminogenemia, and is a severe disorder and the condition primarily discussed in this report.
Congenital type II plasminogen deficiency is a qualitative deficiency with a normal or near normal plasminogen immunoreactive plasminogen level with decreased activity and is also called dysplasminogenemia; type II plasminogen deficiency is not associated with the development of pseudomembranes and is therefore not discussed.
Congenital plasminogen deficiency is a disorder that results in inflamed growths on the mucous membranes, which are the moist tissues that line body openings such as the eyelids and the inside of the mouth.
Development of the growths are usually triggered by infections or injury, but they may also occur spontaneously in the absence of known triggers. The growths may recur after being removed.
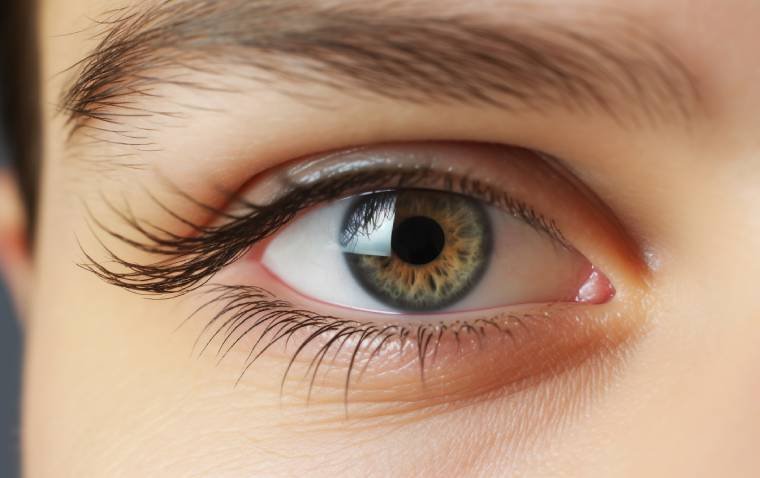
Congenital plasminogen deficiency most often affects the conjunctiva, which are the mucous membranes that protect the white part of the eye (the sclera) and line the eyelids.
A characteristic feature of this disorder is ligneous conjunctivitis, in which a buildup of a protein called fibrin causes inflammation of the conjunctiva (conjunctivitis) and leads to thick, woody (ligneous), inflamed growths that are yellow, white, or red. Ligneous conjunctivitis most often occurs on the inside of the eyelids.
However, in about one-third of cases, ligneous conjunctivitis over the sclera grows onto the cornea, which is the clear covering that protects the colored part of the eye (the iris) and pupil. Such growths can tear the cornea or cause scarring. These corneal problems as well as obstruction by growths inside the eyelid can lead to vision loss.
People with congenital plasminogen deficiency may also develop ligneous growths on other mucous membranes, including the inside of the mouth and the gums; the lining of the nasal cavity; and in females, the vagina.
Growths on the mucous membranes that line the gastrointestinal tract may result in ulcers. The growths may also develop in the windpipe, which can cause life-threatening airway obstruction, especially in children.
In a small number of cases, affected individuals are born with impaired drainage of the fluid that surrounds and protects the brain and spinal cord (the cerebrospinal fluid or CSF), resulting in a buildup of this fluid in the skull (occlusive hydrocephalus). It is unclear how this feature is related to the other signs and symptoms of congenital plasminogen deficiency.
When Left Untreated
When left untreated, LC can result in visual impairment or blindness. Presentation of visual symptoms typically results in ophthalmologists being the first clinicians to evaluate and potentially diagnose patients at symptom onset.
As the disease’s prevalence is currently estimated at 1-2 per one million in the general population, C-PLGD is often misdiagnosed. This is due to both its rarity and the fact that symptoms are often ascribed to other, more common entities.
There exist important knowledge gaps in C-PLGD, with little known of its true incidence, triggers, disease course based upon established severity categories, and overall impact.
Quantifiable information about disease management, the need for surveillance of other potentially affected areas, the burden of disease, and patient experience is limited.
Clinicians currently rely on anecdotal information, personal experience, and a limited number of case studies to develop patient care plans.
No currently licensed specific and efficacious therapy is available to treat C-PLGD, compounding knowledge gaps, and contributing to less than optimal outcomes.
Teams at the Indiana Hemophilia and Thrombosis Center (IHTC), the Fondazione Angelo Bianchi Bonomi, IRCCS Ca’Granda Ospedale Maggiore Policlinico of Milan (IRCCS), and the University of Milan (UNIMI) are working to address these needs through the first-ever collaborative natural history study of C-PLGD, titled “Hypoplasminogenemia: An International RetroSpecTive and PrOspective CohorRt StudY,” also known as “HISTORY.”
The results of this research effort could have important implications for ophthalmologists in the years ahead.
Current Coagulation Disorder Data Collection
There are currently two main repositories for data from patients with rare coagulation disorders, led by the American Thrombosis and Hemostasis Network (ATHN) and the European Network of Rare Bleeding Disorders (EN-RBD).
While these organizations have made great strides in collecting data, a few issues have limited its applicability. Neither program specifically addresses C-PLGD.
The ATHN, in conjunction with the Centers for Disease Control, has accumulated a data repository with more than 80,000 participants, but it is a US-based program, making data regarding treatment and outcomes of included populations less applicable to patients living in other countries with different medical resources and populations.
The EN-RBD created the Rare Bleeding Disorders Database in 2004 to organize clinical, genetic, and treatment data surrounding rare bleeding disorders beyond classical hemophilia A and B. Early data from the study was collected from 592 patients based in 11 European countries.
This data allowed for the development of a new classification system for the severity of bleeding symptoms associated with these disorders.
Since its inception, the database (later renamed the Prospective Rare Bleeding Disorders Database (PRO-RBDD)) has expanded to include prospective data collection from patients at 62 centers throughout the world.
The goal of the expanded study is to support more accurate assessments of the prevalence of specific rare bleeding disorders, in addition to gaining new insight into symptoms, disease burden, and clinical management.
While results have the potential to provide important information related to all rare bleeding disorders, the study does not include C-PLGD.
Making HISTORY
The HISTORY project is the first international study aiming to assess and define the natural history of C-PLGD, Blood Ties Hematology researchers are embarking on the world’s first collaborative natural history study on congenital plasminogen deficiency – and ophthalmologists have a critical role to play By Amy Shapiro define severity categories, and identify triggers of disease manifestations.
Coordinated by IHTC and IRCCS/ UNIMI, it is an extension of PRO-RBDD that will take place over four years – participants provide one year of retrospective data at baseline, followed by three years of prospective data.
Upon enrollment, subjects are evaluated at an in-person visit at a study center to collect retrospective data, perform a physical examination, and obtain biological samples. Participants then have follow-up data collection conducted via telephone every six months until the end of the study, when they are seen in person.
If subjects experience a clinical event suspected to be due or related to pseudomembrane development, or another medical event such as pregnancy, they will be seen in person for evaluation and sample collection.
Researchers will collect data from approximately 500 participants, including up to 100 C-PLGD patients and their first-degree family members.
The study will evaluate several elements, including participants’ overall health, age at diagnosis, the reason for original C-PLGD screening, phenotypes, and genotypes.
Participants with confirmed C-PLGD diagnoses will be classified as asymptomatic, intermittently symptomatic, or continuously symptomatic and their data will be considered concerning gender, environmental influences, genetic information, and other laboratory measures.
C-PLGD-related lesions will be observed based on-site, frequency, and duration in addition to the type(s) and duration of treatments.
The study will evaluate laboratory parameters, information surrounding the management of surgical procedures, obstetric data, and any treatment complications. One key aspect of this study will be the collection of data from asymptomatic first-degree family members of C-PLGD patients.
This data will make a significant contribution to C-PLGD literature, as asymptomatic affected patients may not be tested for the disease and heterozygous individuals are not routinely assessed or followed.
Data from affected asymptomatic participants in the study may hold valuable information regarding the triggers of disease manifestations. Also, data from heterozygous family members will provide insight into the relationship between minimal plasminogen activity levels and the natural history of C-PLGD.
Implications of C-PLGD Research In Ophthalmology
Researchers hope to employ data and samples collected to address knowledge gaps in the clinical care of C-PLGD, ultimately establishing a superior standard of care.
Given that LC is a common first presenting symptom of C-PLGD, the data have the potential to provide significant support to ophthalmologists and other clinical eye healthcare providers who currently have very limited information and guidance for care of these patients.
For example, if patients experience LC, there is currently no data available to assess the likelihood that they will develop lesions in other systems – including ones that could be life-threatening – what surveillance methods should be utilized and when, how frequently each person should be monitored, or how long they should receive treatment.
While an ophthalmologist may be able to provide some symptom relief for LC, they may not be able to predict what other specialists the patient will need to see.
The HISTORY study could support the development of consistent and effective standards of care for patients with C-PLGD, including those who present with LC.
Currently, there is no approved therapy for C-PLGD so medications are prescribed off-label, providing inconsistent and less than optimal outcomes.
The HISTORY project aims to collect data in a way that allows for the development and confirmation of disease severity categories, more accurate prediction of the disease’s course, effective identification of specific surveillance needs in subpopulations and the creation of treatment algorithms, more comprehensive recommendations, and best practices to guide disease management.
The HISTORY project also has the potential to help form the foundation for future clinical research through the development of a biorepository and the use of advanced coagulation testing. Researchers can potentially use information collected during the study to evaluate the use of locally performed tests as diagnostic tools.
The development of a disease-specific screening test would allow for assistance in diagnosis and prediction of disease course, facilitating earlier intervention and establishment of a more accurate prevalence rate.
Currently, the diagnosis of C-PLGD is confirmed through analysis of blood plasminogen activity and antigen levels. These tests are not universally available and may not be standardized. Mutation analysis provides further confirmation but also is not always available.
Clinical research in ultra-rare diseases, as exemplified by C-PLGD, is challenged by both patient identification, phenotypic heterogeneity, and ultimately recruitment. The small populations of affected individuals involved in trials make generalizing data difficult.
The data collected and analyzed during this study may assist in identifying patients for future clinical trial eligibility.
While there are currently no approved specific and efficacious treatments available, researchers are exploring the use of a plasminogen concentrate derived and isolated from the blood of healthy donors to treat C-PLGD. The investigational therapy is currently under review by the FDA.
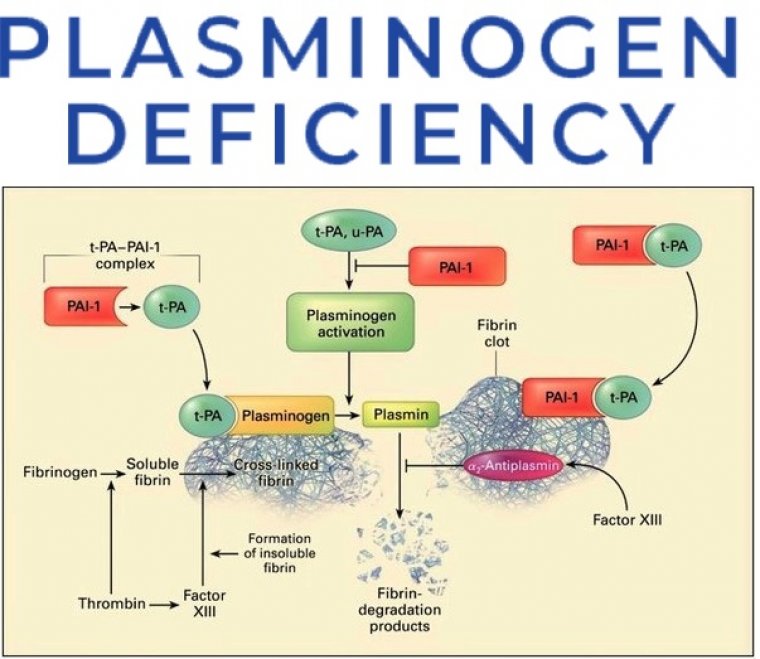
Plasminogen is a key zymogen that plays a fundamental role as the precursor of plasmin, the main enzyme involved in the lysis of blood clots and clearance of fibrin; thus, plasminogen plays a central role in the maintenance of mucous membranes, wound healing, cell migration, tissue remodeling, angiogenesis, and embryogenesis.
One of the most important goals of the HISTORY project is to increase awareness of C-PLGD among ophthalmologists and other medical specialists who may play a central role in patient care. Providing ophthalmologists with new levels of quantif iable information and insight into C-PLGD has the potential to improve time to diagnosis and the ability to advance patients to appropriate care.