
Verana Health has introduced its Qdata Thyroid Eye Disease (TED) module, a new addition to its suite...
read more
LEP Biomedical announced pre-seed funding of €100,000 ($107,000) over two 6-month periods, which the...
read more
SalioGen Therapeutics announced the selection of SGT-1001 as a development candidate for Stargardt d...
read more
Phoenix-Micron announced a new collaboration with ArtiKode Intelligence, a pioneering artificial int...
read more
Ocugen has officially completed dosing in the second cohort of its phase 1/2 ArMaDa trial of OCU410,...
read more
iHealthScreen has been granted a full US patent for its iPredict glaucoma detection model by the US ...
read more
ENTOD Pharmaceuticals, a Mumbai-based company, has achieved a significant milestone in eye care with...
read more
EyeSouth Partners, a prominent eye care-focused management services organization, announced its newe...
read more
In an inspiring leap towards health equity, a new eye health project in Malawi has successfully brou...
read more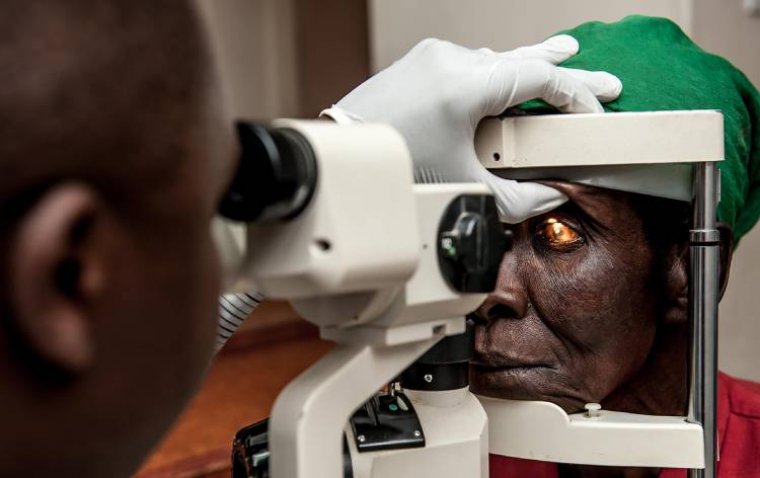
Caplin Steriles Limited (Caplin), a subsidiary of Caplin Point Laboratories Limited, has received fi...
read more
 More
More