Treating Retinal & Optic Nerve Degeneration
More than 1 million nerve fibers make up the optic nerve, which carries visual information. We have one that connects the retina at the rear of each eye to the brain. Loss of vision can occur when an optic nerve is damaged. Where the injury occurs affects the type of visual loss and how severe it is. One or both eyes may be impacted.
Optic nerve problems come in a wide variety of forms, including:
- The main cause of blindness in the US is a collection of conditions known as glaucoma. Glaucoma typically develops as a result of gradual increases in intraocular fluid pressure that harm the visual nerve.
- An inflammation of the optic nerve is called optic neuritis. Infections and immune-related diseases like multiple sclerosis are among the causes. Sometimes there is no known cause.
- The optic nerve is harmed by optic nerve atrophy. Poor blood supply to the eye, illness, trauma, or exposure to hazardous substances are some of the causes.
- Over time, the optic nerve develops pockets of protein and calcium salts called drusen.
Eye exams, ophthalmoscopies (examinations of the back of the eye), and imaging studies may all be used as diagnostic tools for diseases of the optic nerve. Depending on the disorder we have, the treatment will vary. In some cases of optic nerve diseases, we can restore eyesight. Others do not respond to treatment, or it may simply stop further visual loss.
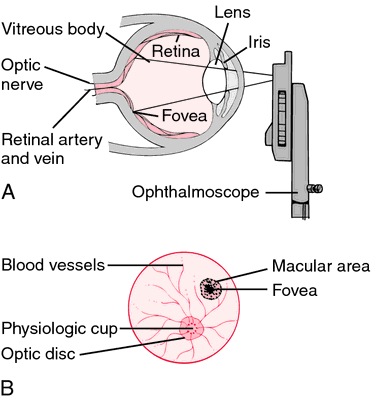
Inherited retinopathies affect one in two thousand persons globally, however there are few effective treatments for retinal degeneration. Glaucoma, which affects 60 million people worldwide and is brought on by the degeneration of retinal ganglion cells (RGCs) and the axon bundles that emanate from them and make up the optic nerve, likewise has no known treatment.
Since LUXTURNA was approved by the FDA in 2017 to treat inherited retinal degeneration brought on by biallelic mutations in RPE65, viral vectors have become a clinically viable treatment option for monogenic retinal illness.
Retinal pigment epithelium (RPE) is now in clinical trials as a cell treatment for treating age-related macular degeneration because to advancements in pluripotent stem cell technology (AMD).
It's time to discuss in vivo reprogramming, a unique treatment strategy that will avoid problems with subretinal cell transplantation and the difficulty of using gene therapy to treat any monogenic illness.
How The Eye Works
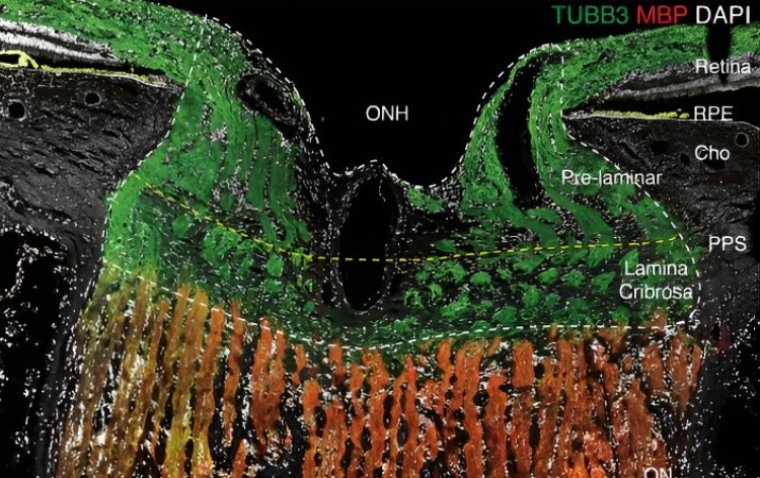
The neural retina's photoreceptors absorb photons and cascade a signal via isomerization of 11-cis retinal and conformational changes in membrane-bound opsin proteins in order to enable vision. The signal is sent to bipolar, horizontal, and then RGCs in the retina as a result of this phototransduction, which results in hyperpolarization and closure of cGMP-gated cation channels. The underlying RPE phagocytoses the photoreceptor outer segments, which are heavily packed with opsins. Retinal cell treatments have an immunological advantage because the RPE monolayer creates a blood-retina barrier. Normally, RGCs are in charge of carrying visual information from photoreceptors to the brain, however it has been shown that a small subpopulation of RGCs is photosensitive and plays a role in the control of circadian rhythms.
The optic nerve, whose axons carry visual information from the retina to the occipital lobe, is made up of RGCs. Rods, one of the two types of photoreceptors, make up up to 95% of all photoreceptors and are in charge of low light and peripheral vision, while cones make up only 5% of all photoreceptors and are in charge of central vision and color perception. Cones are typically present in the fovea and express S/M/L opsins, whereas rods express the rhodopsin protein. In contrast to CRX, which is abundantly expressed in both rods and cones, NRL is a transcription factor that determines the identity of rod cells.
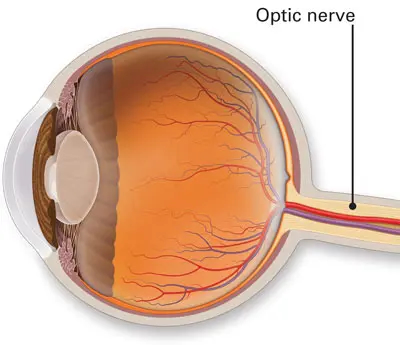
RGCs express ATOH7 and BRN3a/BRN3b transcription factors, whereas MITF is a well-known transcription factor that is mostly expressed in RPE. Data-driven cell conversions can result in the creation of novel cell treatments and in vivo reprogramming strategies to cure retinal degeneration by using the power of transcription factors to govern retinal cell identities and trans-differentiation.
The Retinal and Optic Nerve Degeneration
Let's go over many forms of retinal and optic nerve degeneration and talk about how in vivo reprogramming might be used to treat them. Up to 170 million people worldwide are affected by AMD, the most prevalent kind of retinal degeneration. The degeneration of the RPE and photoreceptor layers has numerous causes, but there are currently no approved treatments that can stop it. Anti-VEGF therapy is one of the treatments for wet AMD because it stops the growth of new blood vessels. RPE generated from human embryonic stem cells (ESC) has undergone phase I/II clinical studies to assess safety and efficacy, but no cell or gene therapy has yet been approved to stop or maybe reverse the progression of AMD.
The diverse disorders known as inherited retinal degenerations cause a progressive loss of photoreceptors and visual acuity. Up to 1 in 4,000 people in the US and around the world are affected by retinal illnesses known as retinitis pigmentosa (RP), which are phenotypically and genetically diverse inherited retinal disorders. Rhodopsin, RPE65, USH2A, PDE6A, and PDE6B are only a few of the more than 50 photoreceptor-related genes known to be mutated in RP, which is often characterized by these mutations. Rhodopsin mutations are the most common cause of autosomal dominant RP, accounting for less than 18% of cases.
Autosomal recessive or X-linked recessive RP, as in the case of RP2, are less frequent types. Cone-rod dystrophies are thought to affect 1 in 30,000–40,000 individuals, and 30–60% of autosomal recessive cases are thought to be caused by ABCA4 mutations. Up to 50% of instances of autosomal dominant cone-rod dystrophies are caused by mutations in the GUCY2D and CRX genes. While cell therapies also have the potential to treat all RP subtypes, gene therapies have already demonstrated efficacy in treating monogenic RP, and retinal gene therapy is generating significant commercial interest.
However, since photoreceptor integration and synaptic connections are more likely with in vivo reprogramming than with cell transplantation, this method may be more effective in treating late-stage retinal degeneration. This is due to the fact that it makes use of Müller glia that are already there, which are close to photoreceptors and have gap junctions with retinal neurons. The second most prevalent cause of blindness in the world, glaucoma includes degeneration of the optic nerve that links the retina to the brain and accounts for 12% of global blindness.
It is characterized by RGC mortality, which is most frequently brought on by high intraocular pressure, which causes the lamina cribrosa pores at the optic nerve head to distort. The RGCs leave the retina through a pore-filled extracellular matrix called the lamina cribrosa, and their axons develop into the optic nerve. The most frequent genetic causes of primary open-angle glaucoma are mutations in the MYOC gene. Cell and gene therapies hold the greatest promise for restoring the damage glaucoma has done to eyesight. Current treatments concentrate on decreasing intraocular pressure to halt the progression of glaucoma.
Retinal Degeneration & Cell Therapy
Why has it been shown that cell and gene therapies work well for the eye? The transparent cornea and lens make it easily accessible, immune-privileged, and well-suited for clinical imaging. Imaging methods like optical coherence tomography and fluorescence adaptive optics scanning light ophthalmoscopy imaging, for instance, can be used to therapeutically examine the retina.
In this manner, the study can continue while engrafted cells and retinal structure are observed after therapy. As a result, it is simple to evaluate the efficacy and safety of cell and gene therapies over time in both animal and human clinical research. As a source of several retinal cell types for study and application, ESCs and induced pluripotent stem cells (iPSCs) are clinically useful. These cells were used to create retinal organoids, which have greatly advanced our understanding of how the retina develops and served as a useful tool for research on photoreceptor isolation and transplantation.
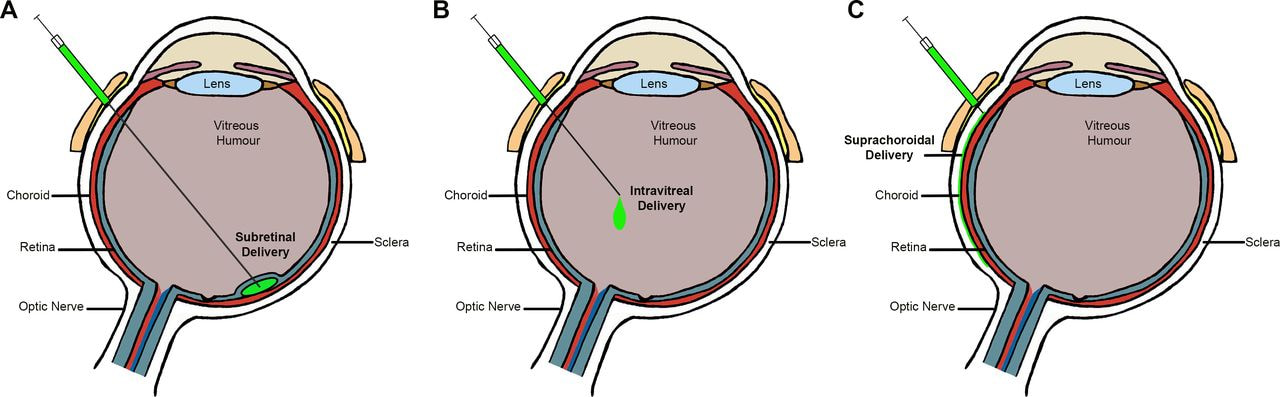
Several clinical trials have been conducted to evaluate the efficacy and safety of RPE produced from ESCs/iPSCs for the treatment of AMD. RPE can be formed spontaneously from iPSCs by removing FGF2 from pluripotency media, or it can be made more effectively by adding small molecules such nicotinamide and activin A. Through subretinal implantation, RPE can be delivered as a cell solution or as a cell sheet. Using transcription factors like MITF, some research have also attempted to transdifferentiate fibroblasts to RPE for clinical transplantation.
RPE transplantation to cure AMD has advanced to phase II of clinical trials, and many delivery methods for RPE made from pluripotent stem cells are now being researched. Since cell treatments have the ability to treat all RP mutation variations by repairing photoreceptors, they are a promising approach for addressing hereditary retinal illnesses. The host mutation would not be transferred by allogeneic therapy, and the RPE forms a blood-retina barrier that confers immunological privilege. Recent attempts have concentrated on producing Müller glia from organoids that can be isolated for sub-retinal transplantation and enhancement of RGC function, as well as increasing cone photoreceptor density in organoids so that they may be isolated for transplantation.
Fetal retinal progenitor cells (RPCs) have showed promising results in a phase IIb clinical trial for the treatment of RP, and they are currently being studied in phase I/IIa clinical trials. The possibility of treating hereditary retinopathies with RPC-based cell therapy remains enticing.
RGCs are produced from iPSCs and retinal organoids using a number of documented techniques, and they appear early in the development of the retina. Due to the length of the axons needed to connect the retina to the visual cortex at the back of the brain, their use as a cell therapy is still challenging. The high intraocular pressure and lamina cribrosa abnormalities must also be fixed for RGC cell therapies to be effective in order to stop additional harm to the donor RGCs. Since there are limited possibilities for gene therapy, RGC therapy offers a very genuine chance of optic nerve regeneration despite its difficulties.
Retinal Degeneration & Gene Therapy
Strategies for gene therapy may target pathways involved in inflammation or visual transduction, or they may try to suppress, replace, or repair faulty genes that are the root cause of hereditary retinal illnesses. Adeno-associated viruses (AAVs) are non-pathogenic and have been proven to successfully target retinal cells after subretinal injection, including photoreceptors and RPE. Recently, LUXTURNA was given FDA approval to use AAV vectors to treat inherited retinal diseases. It is used specifically to treat Leber's congenital amaurosis (LCA) or RP brought on by verified biallelic RPE65 mutations.
The approval has generated a lot of interest in creating AAV gene treatments for inherited retinal diseases. efforts have concentrated on generating can be isolated from organoids for sub-retinal splantation and improvement of RGC function, while cone photoreceptor density can be increased in organoids so they can be isolated for transplantation. The retinal pigment epithelium forms the blood-retina barrier that helps to prevent this.
Lentivirus has been used to deliver these genes in order to evaluate their effectiveness and safety. Although the level of expression may not be sufficient to produce a therapeutic impact, a dual vector strategy has recently been employed to restore the function of ABCA4 in homozygous null mice. Antisense oligonucleotides for transitory expression, such as CEP290 for LCA and QR-421a for LCA involving USH2A, are another method for delivering gene therapy. Recently, retinal organoids have been used to model RP2 mutation-induced X-linked RP.
In RP2-mutated organoids from patients, gene therapy administered by AAVs can treat photoreceptor degeneration and thinned outer nuclear layer. AAVs have also been used to target RPGR as part of a gene therapy strategy to treat X-linked RP. Microglia have been activated with TGF-1 using AAVs in a mouse model of retinal degeneration to protect cone photoreceptors. TGF-1 was delivered via AAV to three mice models of RP containing distinct mutations, saving degenerating cones.
The findings imply that TGF-1 may help patients with neurodegeneration and can protect vision. An innovative method of gene therapy for AMD prevents inflammation brought on by the complement system. Phase I/II trials to evaluate the safety and effectiveness of this gene therapy in treating dry AMD in people began in January 2019; to date, there have been no safety problems. In this case, AAVs are employed to deliver a proprietary protein after vitrectomy.
There is also proof that gene treatments may benefit glaucoma patients by employing CRISPR-Cas9 to target aquaporin-1 in the ciliary body and lower intraocular pressure. According to additional research, the CRISPR-Cas9 method of gene editing MYOC has been successful in reducing intraocular pressure in glaucomatous mouse eyes.
After retinal degeneration, cell and gene therapies may be able to restore vision. However, these treatments are still difficult to distribute and integrate, and there aren't many approved advanced therapeutic medicines. Here, we assess Müller glia in vivo reprogramming as a novel strategy for treating late-stage retinal degeneration.
Reprogramming
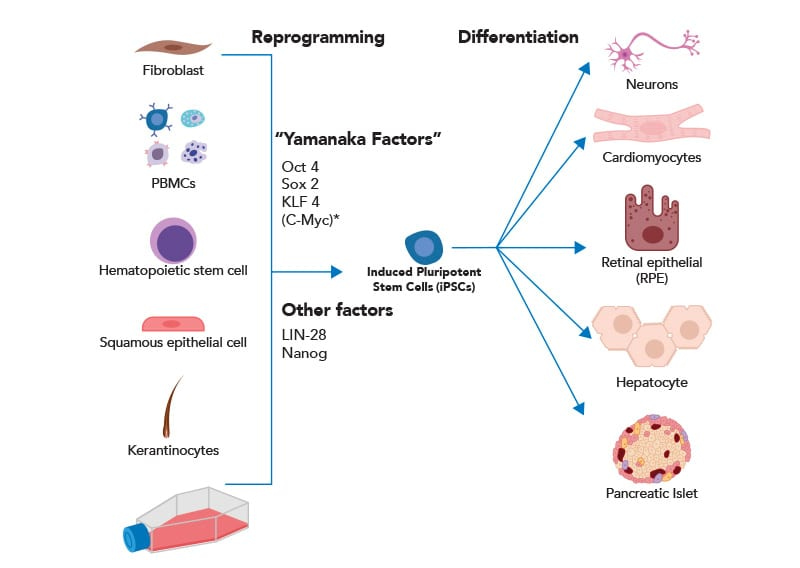
Since it was discovered that the transcription factors OCT4, KLF4, SOX2, and cMYC may convert somatic cells into induced pluripotent stem cells, there has been a lot of interest in using direct reprogramming to create new cell therapies. The transformation of fibroblasts into neurons and cardiomyocytes are two examples of direct reprogramming; it is challenging to foresee the necessary reprogramming elements for cell conversion, though. Big-data strategies, like the algorithm we created, can use RNA sequencing to forecast the transcription factors necessary to change any cell type.
The best transcription factors are then employed to convert various cell types in vitro and in vivo. Transcription factors are then graded according to their impact on the gene expression profile of the target cells. There is precedent for reprogramming retinal cell types, including Müller glia in vivo and fibroblasts to rods.
Thus, a novel and intriguing therapeutic strategy for treating photoreceptor degeneration involves the direct administration of transcription factors for in vivo reprogramming of retinal cell types. As the retina's main support cell and the first to respond to injury, Müller glia represent the best possible source in the retina for in vivo conversion to photoreceptors.
Müller glia cells have been found to regenerate other retinal cell types in the zebrafish retina after injury, which shows they have progenitor capabilities. It was discovered that Müller glia had a role in the regeneration of neurons by up-regulating the ASCL1 gene.
As a result of epigenetic repression, Müller glia in rats are not involved in retinal regeneration; nevertheless, ASCL1 overexpression and the application of the histone deacetylase inhibitor trichostatin A have been shown to stimulate Müller glia to produce neurons. By fusing Müller glia with hematopoietic progenitors, which has been demonstrated to promote the cells' conversion to photoreceptor precursors, Müller glia can also be reprogrammed. More recently, Müller glia were converted into rod photoreceptors in Gnat1rd17Gnat2cpfl3 double mutant mice via the transcription factors OTX2, CRX, and NRL.
In order to reprogram Müller glia in mice lacking functional photoreceptors to function as rod photoreceptors, this study employed beta-catenin to promote Müller glial growth. Overall, the possibility to stop retinal degeneration by transforming Müller glia into photoreceptors shows great promise. Due to its proximity to photoreceptors in the outer nuclear layer and in vitro demonstration of its stem cell capabilities, RPE is another option for photoreceptor regeneration. RPE, on the other hand, is a quiescent monolayer, making the conversion of RPE cells into photoreceptors paradoxical given their crucial function in maintaining photoreceptor homeostasis.
The regeneration of RGCs and the therapy of glaucoma are two additional attractive applications of in vivo reprogramming. Overexpressing the established transcription factors Atoh7, Brn3B, Sox4, Sox11, and Isl1 in amacrine interneurons results in the conversion of these neurons to RGCs. Additionally, there is proof that the transcription factors Ascl1, Brn3b, and Isl1 can produce RGCs from murine fibroblasts. Br Despite not being necessary for the formation of RGCs, overexpression of KLF4 has also been demonstrated to trigger their regeneration in vivo.
We will be able to forecast the most effective transcription factor combinations for reprogramming in vivo, generating cells for therapy, and converting retinal cell types utilizing a systematic data-driven strategy.