
The U.S. Department of Justice (DOJ) has initiated a lawsuit against Regeneron Pharmaceuticals under...
read more
Glaukos has announced the assignment of a unique, permanent J-code (J7355) by CMS for its iDose TR (...
read more
A recent study has introduced an innovative approach to treating retinal detachment through an artif...
read more
Waterloo scientists have developed groundbreaking contact lenses that both protect corneal wounds an...
read more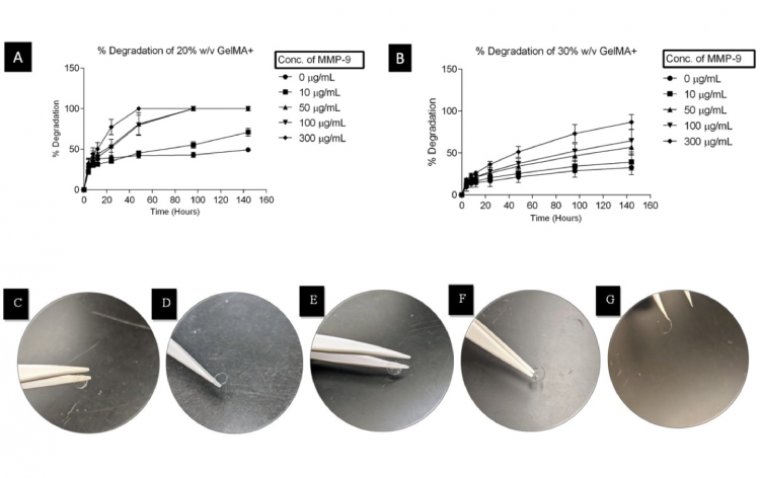
Research teams, under the guidance of a faculty member from Purdue University’s College of Engineeri...
read more
In an innovative stride toward combating age-related macular degeneration (AMD), Oculogenex, Inc. ha...
read more
Researchers have unveiled an artificial intelligence (AI) technology capable of independently detect...
read more
AcuSurgical announced the successful completion of the world’s first retinal surgery performed using...
read more
The U.S. Food and Drug Administration (FDA) has approved clobetasol propionate ophthalmic suspension...
read more
Nethradhama Super Speciality Eye Hospitals achieved a remarkable feat in pediatric eye care with the...
read more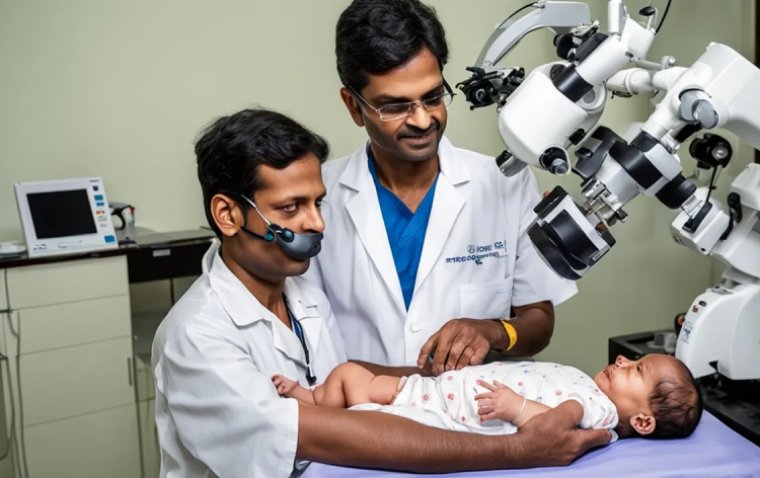
 More
More
